


English
Request a Quote



Using use human or animal cells, tissues and body fluids as raw materials, biological products may introduce human or animal-derived raw materials and auxiliary substances during preparation and formulation stages. This poses huge potential risk of viral contamination.
To effectively mitigate the risk of viral contamination in biological products, regulatory authorities both domestically and internationally have enacted relevant regulations, mandating the adoption of effective virus inactivation and removal steps in the production processes of biological products to ensure the safety of final products.
Viral contamination is one of the significant threats to the safety of therapeutic biological products. Historically, a lack of comprehensive awareness of the risks combined with limitations in viral detection technology and inactivation/removal methods, serious viral contamination incidents occurred in the production process of blood-derived biological products and those produced by engineered cells and bacteria. Refer to table 1 for more details.
Table 1 Viral contamination cases in history
| Year | Biologics | Viral contamination |
|---|---|---|
| 1940s | Yellow Fever Vaccine | Avian Leukosis Virus, Hepatitis B Virus |
| 1950s | Polio Vaccine | SV40 |
| 1980s | Human Growth Hormone | Creutzfeld-Jakob |
| 1980s | Blood-derived Products | HIV, HBV, HCV |
| 1990s | Blood-derived Products | HAV, HCV |
| 1990s | Monoclonal Antibody | LCMV, MVM |
| 2009 | Recombinant Protein | Vesivirus |
| 2010 | Vaccine | Porcine Circovirus |
As virus safety control in biologic products continuously improves, a series regulations and technical specifications have been issued for the safety control and assessment of biologics based on the long-term accumulated experience of responsible regulatory authorities worldwide. The followings are relevant regulations referred in this article:
1) General Principles for Technical review of viral safety evaluation of biological tissue extraction products and Eukaryotic cell expression products, Center for Drug Evaluation of NMPA, 2005.12
2) Technical methods and validation guidelines for viral clearance/inactivation of blood products, NMPA, 2002.05
3) ICH Q5A(R1):Viral safety evaluation of biotechnological products derived from human or animal cell lines, ICH, 1999
4) Chinese Pharmacopoeia(three books), General Principles of Biological Products - Biological products virus safety control, 2020 version
5) Technical Report No.83-Virus Contamination in Bio-manufacturing: Risk Mitigation, Preparedness, and Response. PDA. 2019
6) Guidance for Industry -Process Validation: General Principles and Practices. FDA. 2011
According to the latest requirements in the 2020 edition of the Chinese Pharmacopoeia, special attention must be paid to the following categories for viral safety in biological products, appropriate preventive measures must been taken:
1. Human blood products:Raw plasma is directly sourced from humans, which poses high risk for viral infection. For these products, focus should be given to virus risk controls of human plasma sources and ensure effective virus removal capabilities in production process. Specifically, for clotting factor products, two different steps(i.e. S/D method and nano-filtration) shall be adopted for virus removal/inactivation.
For Immunoglobulin products, it is recommended to use two different virus removal/inactivation methods such as low pH incubation and nano-filtration. For albumin products, low-temperature ethanol process in downstream purification has already provides some virus inactivation effect, which can be combined with pasteurization for further safety control. In addition, virus safety tracing of marketed products can also be required if necessary.
2. Recombinant therapeutic biological products: focus must be given to the source control of engineered cell substrates, engineered bacteria, raw materials and excipients, while ensuring the strong viral clearance capacities of production process.
3. Products sourced from animal body fluids/tissues: special consideration should be given to controlling the risk of animal viruses in the starting materials, especially zoonotic viruses, while ensuring the effective viral clearance in production process. If necessary, testing for viral contamination should be conducted to ensure their safety.
The sources of upstream contamination in biological products include both endogenous and exogenous viral contamination. Endogenous viral contamination sources include engineered cells, which may originate from human tissues or organs (e.g. animal tissues or organs, plant tissues, or other sources). Once these engineered cells are contaminated, viruses will be introduced to downstream processes and become a source of viral contamination.
Exogenous contamination sources mainly include logistics control, personnel control, facility environment and process monitoring. Take the biological products derived from engineered cells as an example, ensuring a contamination-free cell bank is crucial, which means regulatory authorities have clear and stringent requirements for contamination testing of cell banks.
Cell bank identification requires comprehensive validation from the Master Cell Bank (MCB), Working Cell Bank (WCB), to final production cells (EOPC/CAL). For the release of unprocessed harvest batches, each production batch of harvested cells/unprocessed cell harvest fluid (UPB) must undergo testing for exogenous viruses and mycoplasma. The analysis and risk control of virus contamination in biological products from other sources can use this as a reference.
In a GLP laboratory environment, a certain amount of model virus is intentionally added (i.e., “spiked”) into unprocessed harvest fluids or intermediates at various production stages. This practice aims to quantitatively assess the effectiveness of virus inactivation/removal process by measuring the reduction levels of viruses during the process and thus determining the overall level of virus reduction that the process can achieve to ensure product safety.
Virus inactivation/removal processes are core aspects of ensuring the safety of biological drugs. According to strict regulations from regulatory authorities, the production processes of biological products must include steps that can effectively inactivate/remove viruses. Detailed virus clearance study reports must be provided when submitting an IND/BLA application. After the product is marketed, based on actual needs and changes in the production process, a re-validation on process effectiveness shall be carried out (see Figure 1).

Fig.1 validation timing of viral clearance
The guidelines specify that a typical validation study should select indicator viruses that comprehensively cover various types. Specific requirements include: 1) include single-stranded and double-stranded RNA and DNA viruses, enveloped and non-enveloped viruses, viruses with strong and weak resistance, as well as viruses of varying particle sizes. In light of relevant regulatory requirements, the selection of model viruses should adhere to the following six principles: 1) both enveloped and non-enveloped viruses should be included; 2) DNA viruses and RNA viruses should be covered; 3) both large and small particle viruses should be included; 4) viruses with similar or close potential contamination risks should be selected; 5) viruses that are highly resistant to the selected inactivation/removal methods should be considered; 6) alternatives that are safe for laboratory personnel should be prioritized.
Regulations further require that the selected indicator viruses should be as non-pathogenic to humans as possible, similar to the potential contaminating viruses, easily cultivable in vitro, and suitable for the specific product characteristics and process features, with resistance to the verification process. Based on these requirements, model viruses used for virus clearance studies can be categorized into three types: relevant viruses (e.g., human viruses associated with blood products), specific model viruses (e.g., MuLV and MVM viruses from CHO expression systems), and non-specific model viruses (e.g., PRV and Reo3 viruses from CHO expression systems). In practice, priority should be given to selecting viruses closely related to potential contaminating viruses to ensure the validity and reliability of the verification study.
Tables 2, 3, and 4 list common model viruses and their main characteristics in CHO expression biological products, baculovirus/SF9 expression systems and blood products, respectively.
Table 2 Model viruses can be selected in CHO expression system
| Virus | Genome | Envelope | Size (nm) | Family | Shape | Resistance |
|---|---|---|---|---|---|---|
| MVM | ssDNA | N | 20~25 | parvovirus | icosahedron | High |
| MuLV | ssRNA | Y | 80~110 | retrovirus | Spherical | Low |
| PRV | dsDNA | Y | 120~200 | herpes virus | Spherical | Intermediate |
| Reo3 | dsRNA | N | 60~80 | reovirus | Spherical | Intermediate |
Table 3. Model viruses can be selected in baculovirus/SF9 expression system
| Virus | Genome | Envelope | Size (nm) | Family | Shape | Resistance |
|---|---|---|---|---|---|---|
| MVM | ssDNA | N | 20~25 | parvovirus | icosahedron | High |
| MuLV | ssRNA | Y | 80~110 | retrovirus | Spherical | Low |
| VSV | ssRNA | Y | 70×150 | rhabdovirus | Bullet-shaped | Intermediate |
| Baculovirus | dsDNA | Y | 45×275 | baculovirus | Rod-shaped | Intermediate |
Table 4 Model viruses can be selected in blood-derived products
| Virus | Genome | Envelope | Size (nm) | Indicator virus |
|---|---|---|---|---|
| HIV | dsRNA | Y | 80~100 | HIV |
| HCV | ssRNA | Y | 40~60 |
BVDV SBV |
| HBV | pdsDNA | Y | 45 |
DHBV, PRV |
| HAV | ssRNA | N | 27 |
HAV, poliovirus, EMCV |
| B19 | ssDNA | N | 20 | CPV, PPV |
The regulations have certain parts concerning the evaluation of the effectiveness of virus inactivation/removal criteria for measuring the virus clearance effects in process steps. Specifically, if the process steps in a validation study can reduce indicator virus quantity by 4 log reduction values (LRV) or more, it is considered effective.
For process steps with removal/inactivation effects between 1 and 4 LRV, it is deemed generally effective. However, when the removal/inactivation effect of a process step is less than 1 LRV, it is regarded as negligible. It is important to note that these negligible process steps will not be counted in the overall virus inactivation/removal LRV total during the assessment, as this may otherwise lead to an overestimation of the actual capacity of viral clearance. These regulations provide clear assessment standards for the effectiveness and reliability of virus inactivation/removal processes.
When selecting and designing viral clearance processes, a series of principles and requirements must be strictly followed. First, the selected process must include at least one effective non-enveloped virus clearance step, which is the base for ensuring viral clearance effectiveness. Additionally, the process must include at least two complementary and effective process steps to enhance the comprehensiveness and reliability of viral clearance.
When designing process schemes, further detailed requirements are stipulated. First, virus removal/inactivation validation studies for dual claim must comply with GLP (Good Laboratory Practices) to ensure the scientific design and accuracy of the research. Secondly, analyze parameters affecting the virus removal/inactivation effectiveness, including mechanical parameters and physicochemical parameters, so as to determine variability limit for these parameters, typically by validating the worst-case conditions to ensure process stability and adaptability.
Furthermore, the kinetics of virus inactivation is also essential. This includes analysis of virus inactivation rates and inactivation curves, which facilitate deeper insights into the dynamic processes of virus inactivation. Moreover, in order to more accurately assess virus clearance effectiveness, the titer of the indicator viruses should be as high as possible to simulate the worst-case virus load.
Finally, when adding viruses for validation, the volume ratio of the virus to the sample being validated should be controlled within a certain range, generally not exceeding 1:9, to avoid affecting the accuracy of the validation results due to excessively high or low virus concentration.
In summary, by adhering to the above-mentioned principles and requirements, a scientific, effective and reliable viral clearance process can be designed. Refer to table 5 for commonly-used viral inactivation/removal processes.
Table 5 viral inactivation/removal processes
| Process | Indicator virus | Advantages/disadvantages | Product |
|---|---|---|---|
| Pasteurization | HIV/HAV, etc. |
Classic method, mild condition. Long processing time, no complete inactivation for some viruses |
Blood-derived products |
| Dry Heat Method | PRV ,PPV,MVM,EMCV, etc. |
Effective to enveloped and non-enveloped viruses, reliability Vigorous conditions, high temperature might affect product stability |
Blood-derived products |
| Solvent/Detergent Method(S/D) | PRV,MuLV,VSV, etc. |
High inactivation effectiveness on enveloped virus, high reliability. Vigorous condition, poor effectiveness on non-enveloped viruses, extra steps required to add S/D agents (cytotoxicity) |
Blood-derived products. Animal tissue extract. Eukaryotic cell expression products. mAb |
| Low pH Inactivation Method | PRV,MuLV,VSV, etc. |
Mild condition, good effectiveness for enveloped virus inactivation. Poor effectiveness on non-enveloped viruses |
Blood-derived products. Eukaryotic cell expression products. mAb |
| Nano-filtration Membrane Filtration | MuLV,MVM,PRV,PPV, Reo3, etc. |
Mild condition, good viral clearance effectiveness for enveloped/non-enveloped viruses. High cost, risk for flow-through, necessity to do through check-up, high requirements towards indicator viruses |
Blood-derived products. Eukaryotic cell expression products. mAb |
| Chromatographic method | MuLV,MVM, PRV,PPV, Reo3, etc. |
Mild condition, existing process for purification of antibody and protein, AEX resin has better performance. Validation is essential in judge viral clearance performance, big difference in performance among different resins. |
Eukaryotic cell expression products. mAb |
Pasteurization is a method that involves continuously heating protein solutions at 60°C for at least 10 hours. The principle is that prolonged high temperatures denature viral proteins, thereby inhibiting the replication of viral genetic material, ultimately rendering the virus non-infectious. Pasteurization is effective against both enveloped and non-enveloped viruses. The method is commonly used in the virus inactivation for blood products including albumin, clotting factors, immunoglobulins, and protease inhibitors.
Dry Heat Method involves treating the product at 80°C for 72 hours, which can inactivate viruses such as HBV, HCV, HIV, and HAV. It is important to consider the moisture content of the product, as well as its composition (such as proteins, sugars, salts, and amino acids), which can influence the virus inactivation effect. The allowable discrepancies in various parameters between product vials should be determined, and dry heat ovens used for virus inactivation should be validated at least every six months. This method is suitable for lyophilized formulations and clotting factor products.
Solvent/Detergent Method (S/D) utilizes a mixture of organic solvents and non-ionic surfactants to disrupt the lipid membranes of enveloped viruses. Typically, a combination of tri-n-butyl phosphate (TNBP) and non-ionic detergents (Triton X-100 or PS80) is chosen. The S/D method is primarily used for inactivating enveloped viruses in clotting factor products, protease inhibitors, and human immunoglobulins.
Since the S/D method introduces organic solvents, it is necessary to remove the added S/D agents during subsequent processing. As shown in Figure 2, use cell culture solution (HCCF) harvested from mAb1 and mAb2 culture to assess the effects of temperature and Triton X-100 concentration on MuLV inactivation. For both monoclonal antibodies, the inactivation rate at 12°C was slower than at 20°C, indicating that lower temperatures and shorter times represent the worst-case scenario; comparisons of Figures A and B indicate that there is no difference in the inactivation effects of 0.3% and 0.5% Triton X-100 on MuLV.
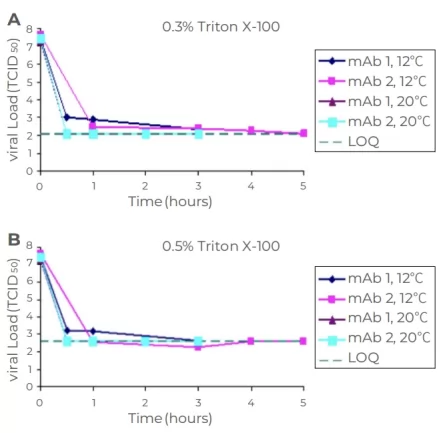
Fig.2 Genentech analysis: effect of temperature (A),time (B) and Triton X 100 concentration on X-MuLV inactivation
Low pH Inactivation Method targets enveloped viruses and requires stable sample under low pH conditions. The low pH inactivation method is commonly used for therapeutic biological products expressed and secreted by recombinant or hybrid animal eukaryotic cells post-culture and those directly extracted and purified from animal tissue raw materials.for viral inactivation effectiveness, lower the pH, higher incubation temperature, longer incubation time and lower sample concentration, all contribute to better inactivation effect.
Nano-filtration Membrane Filtration is currently the most reliable virus removal technology, primarily based on size exclusion, utilizing the difference in size between viruses and proteins. Proteins smaller than the average pore size pass through the membrane, while viruses larger than the average pore size are retained. This method can effectively remove various viruses (both enveloped and non-enveloped). Using membranes with an average pore size of around 20 nm, the
Chromatographic method uses affinity as well as other interaction disparity between viruses and target molecules to achieve fraction isolation when contacting resin. Chromatographic method consists of affinity chromatography, anion exchange chromatography, cation exchange chromatography, mixed-mode chromatography and so one.
Table 6 listed different chromatography methods and their viral clearance results. When using chromatographic method in removal of virus, chromatography types and resin using times(new or previously-used resin) play vital roles in final viral clearance results. Generally speaking, resin using times(new or previously-used resin) will not contribute to the viral clearance ability of resin.
Table 6 Viral clearance results of chromatography technology
| Resin type | Viral clearance (log10) | Recommended resin |
|---|---|---|
|
Affinity chromatography |
2~4 |
AT Protein A Diamond Plus Novo-A Diamond Extrem A Diamond AT Protein A Diamond Ultra |
|
Anion exchange chromatography |
4~5 |
Diamond Q Q Bestarose FF |
|
Cation exchange chromatography |
2~3 |
Diamond CD-S MegaPoly BXS |
|
Hydrophobic interaction chromatography |
4~5 | Phenyl and Butyl series |
|
Mixed-mode AEX chromatography |
4~5 |
Diamond MIX-A Diamond MIX-A Mustang |
Investigational New Drug (IND Stage) The requirements during the IND stage of the drug application focus mainly on the selection of model viruses, process selection, and process reproducibility. Table 7 lists the requirements for domestic and international blood products as well as antibody/recombinant protein products during the IND submission stage.
Table 7. Viral inactivation/clearance validation requirements for international/domestic IND applications
| Resin type | Viral clearance (log10) | Recommended resin |
|---|---|---|
| Affinity chromatography | 2~4 |
AT Protein A Diamond Plus Novo-A Diamond Extrem A Diamond AT Protein A Diamond Ultra |
| Anion exchange chromatography | 4~5 |
Diamond Q Q Bestarose FF |
| Cation exchange chromatography | 2~3 |
Diamond CD-S MegaPoly BXS |
|
Hydrophobic interaction chromatography |
4~5 | Phenyl and Butyl series |
| Mixed-mode AEX chromatography | 4~5 |
Diamond MIX-A Diamond MIX-A Mustang |
Note: when choosing model viruses for blood-derived products, Vesicular stomatitis virus (VSV) can tolerate wider pH range. VSV can be used as indicator virus in the validation of viral clearance performance in low pH incubation method.
Biologics License Application (BLA Stage) The requirements during the BLA stage of the drug application focus on the selection of indicator viruses, process selection, and process reproducibility, with stricter requirements. Table 8 lists the requirements for domestic and international blood products as well as antibody/recombinant protein products during the BLA submission stage.
Table 8 Viral inactivation/clearance validation requirements for international BLA application
| Viral inactivation |
Domestic blood-derived products |
Domestic antibody/recombinant protein products | International antibody/recombinant protein products | |
|---|---|---|---|---|
| Sample quantity | 3 GMP batches | 1 batch, repeat twice | 1 batch, repeat twice | |
| Model viruses | Low pH method | VSV | X-MuLV,PRV | X-MuLV,PRV |
| Nanofiltration Membrane | PPV | MVM,Reo3,X-MuLV,PRV | MVM,Reo3,X-MuLV,PRV | |
| Chromatography 1 | N/A | MVM,Reo3,X-MuLV,PRV | MVM,Reo3,X-MuLV,PRV | |
| Chromatography 2 | N/A | MVM,Reo3,X-MuLV,PRV | MVM,Reo3,X-MuLV,PRV | |
Since it is impractical to carry out viral clearance validation at process-scale, validation will only be achievable at scale-down. Key parameters and control condition of scale-down model shall be strictly uniform with that of practical production process. As for chromatographic process, affinity chromatography and AEX chromatography showed excellent effectiveness in viral clearance.
Therefore, the two chromatography methods are commonly used in the experiment of viral clearance. Table 9 shows the worst process conditions in the viral clearance validation of affinity chromatography and AEX chromatography resins(FT mode).
Table 9 Process validation requirements
| Process | Worst process condition |
|---|---|
| Affinity chromatography |
Upper limit: column height, sample loading volume, elution conductivity Lower limit: Flow, Ve, elution pH |
| AEX chromatography |
Upper limit: sample loading flow, sample loading binding capacity, sample loading conductivity, washing CV Lower limit: bed height, retention time, sample loading pH |
When evaluating viral clearance effectiveness, cytotoxicity and viral interference are two critical factors that must be considered, as they can severely interfere with the accurate measurement of viral titers. Therefore, before adding viruses to intermediates to study their clearance effectiveness, the primary task is to conduct comprehensive studies on cytotoxicity and viral interference for these intermediates to ensure accurate assessment of actual viral titers in the samples.
Additionally, during the verification phase of virus inactivation/removal processes, high demands are placed on the sensitivity, specificity, stability, and reproducibility of viral detection methods.
In the verification of virus inactivation/removal processes, viral detection methods are required to demonstrate high sensitivity, specificity, good stability and reproducibility. Currently, main viral detection methods include plaque-forming unit (PFU) assay (quantitative), cytopathic effect (TCID50 method) (semi-quantitative), and quantitative nucleic acid methods (qPCR method).
• Plaque-Forming Unit (PFU) Method: Applicable for removal and inactivation processes.
• Cytopathic Effect (TCID50 Method): Applicable for removal and inactivation processes.
• Quantitative Nucleic Acid (qPCR Method): Applicable for removal processes; not suitable for inactivation processes.
For viruses where the initial number can be estimated, such as model retroviruses, Figure 7 provides an example of how to calculate the safety factor. The purpose of this calculation is to determine the probability of a single viral particle entering a single-dose product. An excessive clearance of 4-6 log from downstream viral inactivation/removal processes essentially means that only one dose in 104-106 doses could potentially contain viral particles.

Fig.7 The calculation of safety factor for model retroviruses
To effective ensure the safety of biologics, top priority shall be given to raw material contamination risk and the corresponding detection. Meanwhile, viral clearance ability in production process shall be especially focused. According to relevant laws and regulations, scientifically-approved and appropriate viral inactivation/clearance validation methods shall be drafted so as to more accurately estimate the effectiveness of process.
Additionally, viral contamination risk control and detection shall be performed throughout the whole life cycle of biologics, to bring the viral contamination risk to the lowest possible level and eventually ensure the safety of finished products.
[1] NMPA: General principles for technical review of viral safety assessments of biological tissue extracts and eukaryotic cell expression products, 2005.12
[2] NMPA: Technical methods and validation guidelines for virus removal/inactivation of blood products, 2002.05
[3] ICH. Q5A: Viral safety evaluation of biotechnology products derived from cell lines of human or animal origin. 1999
[4] Chinese Pharmacopoeia three sections 2020: General rules for biological products-risk control for biological products viral safety
[5] Technical Report No. 83. Virus Contamination in Biomanufacturing: Risk Mitigation, Preparedness, and Response. PDA. 2019
[6] FDA:Guidance for Industry -Process Validation: General Principles and Practices. 2011
[7] Cytiva academy: Validation of virus inactivation/removal of biological products
[8] Chinese Pharmacopoeia Commission: Key points of risk control for viral contamination of therapeutic biological products, 2021
[9] George Miesegaes.(2014)Viral Clearance by Traditional Operations With Significant Knowledge Gaps (Session II): Cation Exchange Chromatography (CEX) and Detergent Inactivation. PDA
[10] Shukla, A. Aranha, H. (2015) Viral clearance for biopharmaceutical downstream processes. Pharmaceutical Bioprocessing.


Mar
26


Mar
04

Mar
02
.png)




.png)
.png)
.png)

.png)